Introduction to Antibody-Drug Conjugates
Summary
Chemotherapy has been a staple of cancer treatment for decades but suffers from a narrow therapeutic window.
Antibody-drug conjugates (ADCs) are a form of precision cancer medicine in which cytotoxic drugs are delivered specifically to cancer cells.
In multiple cancer types, contemporary ADCs have demonstrated efficacy greatly exceeding traditional chemotherapy.
The ADC platform appears to have reached a critical maturity threshold enabling widespread adoption.
Introduction
Cancer has always been a difficult illness to address. The fundamental objective of patient care is to kill or remove as much cancerous tissue as possible, while leaving healthy cells intact. This idea is much easier said than done. Since the earliest days of medical oncology, physicians have utilized physical methods for treating cancer patients – surgical removal of tumors is often the first course of action and remains highly effective for many cancer types, particularly in the early stages of disease progression [1]. Unfortunately, tumors are frequently more invasive than what can be discerned by the human eye. While surgery is effective at removing the vast bulk of a tumor, there are often tiny clusters of cancerous cells that have previously detached and migrated elsewhere in the body, so called micro-metastases [2]. These residual cancer cells, while initially very small, eventually grow back over time and lead to disease progression. Moreover, surgery is not always a viable option for all patients. A tumor may be growing in a location too risky for surgical removal, or there are so many cancerous nodes throughout the body that attempting to cut each of them out is not a realistic option [3].
Multiple approaches have been developed to address these issues. One of the first was the use of ionizing radiation to kill cancer cells with a focal beam of high-energy particles. This technique has the advantage of being able to destroy tumors almost anywhere in the body in a non-invasive manner, providing hope in situations where operation is inappropriate [4]. Additionally, radiation can be used in combination with surgery by treating the tumor bed and surrounding tissue after lesion removal, a method intended to kill any remaining cancer cells that were left behind [5]. These methods are still in use today and are some of the most widely practiced treatment paradigms in oncology. Radiation can be highly effective at killing cancerous tissue, and healthy cells that are not directly in the path of the radiation beam are minimally affected [4]. As radiation technology has progressed over the years and more sophisticated equipment is being utilized, radiation beams can be extremely precise in delivering toxic doses to highly focused areas with minimal exposure to non-targeted regions. Unfortunately, this precision is also somewhat responsible for the unavoidable relapse that many patients experience – anything not in the path of radiation is not treated. Thus, only tumors that are large enough to be detected by the human eye, or a surgical wound area in which a tumor was removed, are realistic targets. Any metastatic “seeds” that separated from the parent tumor and migrated elsewhere in the body remain undetected until they too are large enough to be seen. Sadly, by the time a metastatic tumor reaches a volume where it can be spotted, it is already too late for many patients.
Chemotherapy
A systemic therapy – one that is delivered into the bloodstream and circulated throughout the entire body – is an appealing solution for eliminating both established tumors and lingering micro-metastases too small for routine detection. While an intriguing idea, the fundamental issue of selectivity remains. How can one design a chemical compound that destroys cancer cells but leaves healthy cells unaffected? Traditional chemotherapy seeks to accomplish this by exploiting unique characteristics of cancer cells that render them vulnerable to attack in specific ways [6]. Because all cancers are inherently driven by an inability to restrain growth and cell division, some molecular features are shared by virtually every tumor family. In a human adult, most healthy cells divide either very slowly or not at all. Some exceptions to this rule are stem cells in the bone marrow (white and red blood cell production), germ cells (sperm production), and epithelial cells (skin and digestive tract) that are constantly being replaced throughout the lifespan [7]. Thus, exploiting weaknesses present specifically in dividing cells (aka the tumor) it is possible to selectively kill cancer cells while healthy tissue is more resilient to this damage [8]. However, the adult cell types that proliferate under normal conditions are also liable to this chemical insult and result in the familiar toxic side effects of chemotherapy. Namely hair loss, infertility, susceptibility to infection, gastrointestinal issues, and many others. Despite these well-known issues, chemo has been an important pillar of medical oncology for many decades and continues to be so today.
In order for cells to divide, the strands of DNA must be replicated into two copies so that one pair each can be allocated to the newly formed daughter cells [9]. DNA synthesis is executed by the concerted action of many proteins, but the major workhorse is an enzyme called DNA polymerase [10]. Like the name implies, DNA polymerase is an enzyme that physically reads the strands of DNA and makes copies of them through the repeated joining of building block molecules known as nucleotides. Under normal conditions, DNA polymerase glides freely across the DNA and efficiently reads its template to synthesize new genetic material. On occasion, various types of chemical damage may accumulate on the nucleobases of DNA from endogenous or exogenous sources and must be removed by DNA repair enzymes [11]. However, DNA damage is not always fixed properly or in a timely fashion. If DNA polymerase encounters these lesions while synthesizing new strands, it is possible for the polymerase to stall or fall off the DNA template completely [12]. This concept of disrupting DNA synthesis also extends to the impairment of other protein components essential for replication. If the cadre of indispensable replication enzymes cannot all function together properly, DNA synthesis comes to a halt. Disrupt this process enough and you will trigger an irreversible cascade of signaling events that results in a systematic, programmed cell death known as apoptosis [13]. The goal of most chemotherapeutic compounds is to damage DNA or interfere with the synthetic machinery to the point where cells can no longer cope and thus trigger apoptosis [6]. Alternatively, some chemotherapies prevent DNA from correctly partitioning into the newly formed daughter cells after cell division – this too results in programmed cell death.
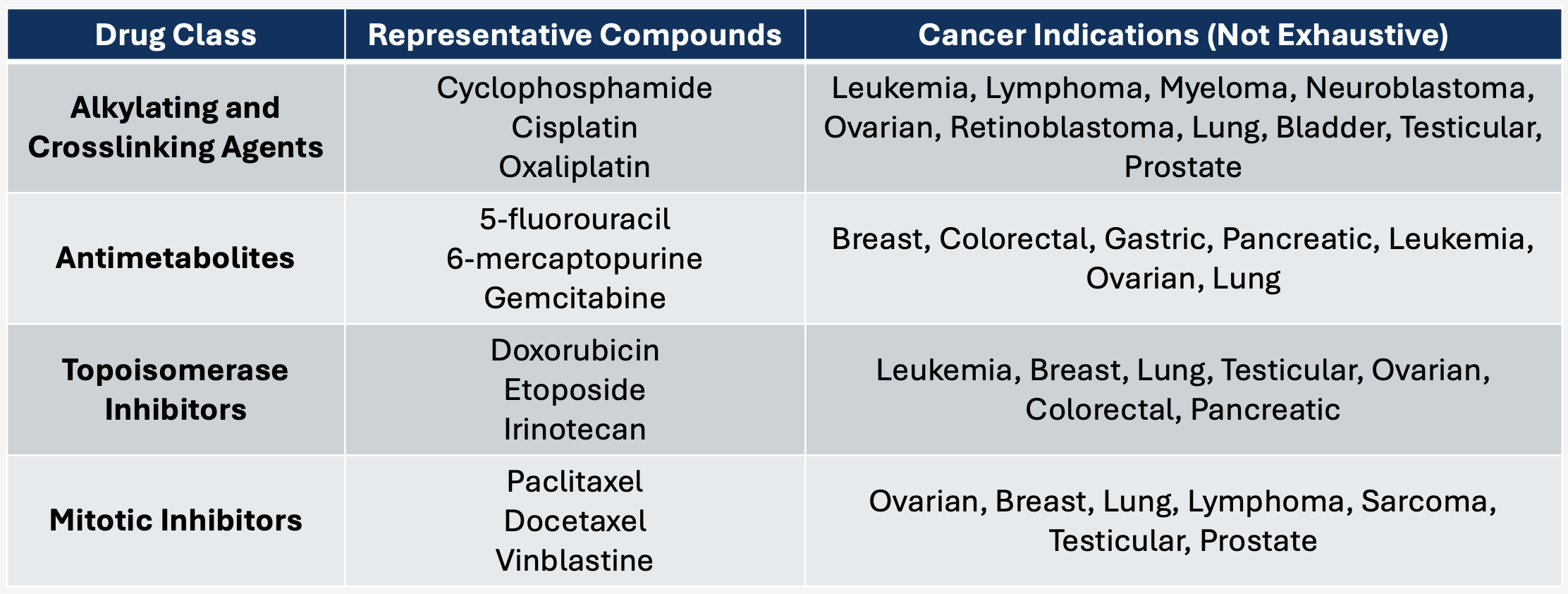
Figure 1. Overview of Chemotherapeutic Medicines. Examples of commonly used medicines and their mechanism of action.
The roots of chemotherapeutic discovery dates to the beginning of the 20th century with German scientist Paul Ehrlich. His pioneering work in the use of animal models for studying infectious disease treatment paved the way for subsequent transplantable tumor models, and these were essential for large-scale screening efforts to identify tumoricidal medications [14]. In 1949, the FDA approved the first chemotherapeutic agent, mechlorethamine, for the treatment of leukemia and various other tumors [15]. At first glance, using mechlorethamine in a therapeutic context seems strange, as related compounds (mustards) were used as bioweapons in WWI. However, a serendipitous discovery occurred during the second World War after an accidental spill of sulfur mustards led to the observation that lymph nodes and bone marrow of poisoned soldiers were destroyed [16]. Intrigued by these observations, scientists tested mechlorethamine in numerous patients as a treatment for lymphoma, a cancer originating from the bone marrow [17]. Researchers were shocked to see that this compound was efficacious as patient’s tumors began responding to treatment. Following this seminal discovery almost 80 years ago, a vast number of chemotherapeutics have been approved for treating a wide variety of cancers. There are multiple distinct classes of chemotherapy [18-29], each with a unique mechanism of exerting their toxic effects:
DNA alkylating and cross-linking agents: These compounds directly react with and covalently modify (damage) the nucleobases of DNA. As described above, excessive amounts of damage obstruct DNA polymerase from functioning properly and inhibit replication. Various classes of DNA repair enzymes exist in the cell to fix damage when it occurs, with different enzyme families evolutionarily specialized for unique types of molecular injury [30]. Alkylating and cross-linking therapies have distinct chemical structures, and as such produce specific types of damage depending on which medicine is utilized. Particular tumor varieties may express higher or lower amounts of DNA repair enzymes, and some are even defective in handling specific classes of damage altogether [31]. Therefore, different cancer types show varying levels of susceptibility to these chemical agents.
Antimetabolites: Drugs of this class mimic the structure of naturally occurring precursor molecules for DNA synthesis (nucleotides) [32]. There are several different ways these compounds exert their toxic effects. Nucleotides are the building blocks of DNA and are physically linked together by DNA polymerase into very long chains that encode our genetic information (chromosomes). Some types of antimetabolites function by inhibiting the enzymes responsible for making these nucleotide building blocks. Without an abundance of nucleotides, DNA cannot be synthesized properly and thus replication is disrupted. Alternatively, some antimetabolites are picked up by DNA polymerase and incorporated into the growing DNA chain. When this occurs, the antimetabolites effectively “cap” the DNA and permanently stop replication from going any further.
Topoisomerase inhibitors: DNA topoisomerases are part of the mixture of auxiliary proteins required for normal DNA synthesis [33]. These enzymes are like molecular scissors – they transiently cut DNA and re-join it together as replication is proceeding. This cutting is essential, as it relieves torsional strain and prevents the unwinding DNA strands from physically spiraling together too tightly (supercoiling). Covalent inhibitors of topoisomerase disrupt this process by irreversibly linking the enzyme to DNA, halting the replication process.
Mitotic inhibitors: During mitosis (cell division), long tube-like protein structures known as microtubules attach themselves to DNA and physically pull chromosomes into each of the newly formed daughter cells. Mitotic inhibitors bind to the key microtubule protein, beta-tubulin, and obstruct the normal de-polymerization process that is required for proper chromosome segregation [34, 35].
Limitations and Innovation
The development of chemotherapy was a major breakthrough in the history of oncology. Combining a systemic treatment with mainstay techniques such as surgery and radiotherapy allowed for meaningful gains in efficacy and patient lifespan [36-39]. Despite the leap forward, there remains much to be desired from this class of medicines. Very few people who undergo chemo are truly cured as most responses are incomplete, and patients inevitably experience disease progression after continued treatment [40]. In the worst scenarios, some fail to see any effect on tumor growth whatsoever. Moreover, systemic toxicity is a major issue with chemotherapy that limits its efficacy. Although cancer cells are more sensitive to these medicines than normal cells, healthy tissue is inevitably damaged in the process. Thus, toxicity in healthy tissues prevents doctors from being able to use sufficiently high doses of chemo to kill every cancer cell. This concept of effectively treating a disease while minimizing undesirable side effects is known as the “therapeutic index” of a compound and is a foundational idea for all of drug development [41].

Figure 2. Schematic of Therapeutic Index. Representative dose-response curves showing the relationship between drug dose, efficacy, and toxicity. The wider the gap between a therapeutic dose of a compound and a toxic dose, the greater the therapeutic index. ADCs were designed to expand the safety and efficacy of chemotherapy
Researchers around the world have sought to identify novel cancer medicines with greater efficacy and a higher therapeutic index, often referred to as “targeted therapies.”[42] Multiple approaches have been developed, each with their own strengths and weaknesses. One major focus has been on understanding how mutated DNA and the encoded mutant proteins initiate cancer cell growth and replication. In many cases, these mutations result in a perpetually active form of a protein that drives unrestrained cellular division (oncoprotein). Since these oncoproteins only exist in malignant cells, inhibiting them with small molecules has shown compelling efficacy and safety in a wide range of tumor indications [43]. However, genetic mutations tend to be highly cancer-type specific and therefore have necessitated the development of many different drugs tailored for unique patient populations. This poses serious issues for rare genetic mutations with a low prevalence, as economic considerations surrounding the end market size may impede project viability. Moreover, the highly specific nature of these drugs can allow for tumor adaptations and resistance over time through compensatory protein mutations [44].
Another incredibly successful approach has been the advent of immune therapy, in which a patient’s own immune system is harnessed to attack and destroy cancer cells [45]. A very common mechanism by which tumors escape targeting by immune cells is by expressing cell-surface ligands that engage receptors known as “checkpoint” proteins on cytotoxic T-lymphocytes (CTLs). These checkpoint proteins, when triggered, effectively halt CTLs from mounting a lethal attack and thus protect the tumor from immune-mediated destruction. Therapeutic antibodies, most notably those targeting PD-1 or PD-L1, stop tumors from being able to activate these checkpoint pathways and empowers CTLs to proficiently recognize and kill cancer cells, a phenomenon known as “de-repression.” Antibodies blocking PD-1 have revolutionized the treatment of many cancer types and are often used in combination with traditional radiotherapy or chemotherapy to enhance efficacy [46]. While checkpoint blockade performs well in a wide variety of cancers, numerous so-called immunologically “cold” tumor varieties do not respond to this treatment [47]. Moreover, some patients experience autoimmune reactions that can damage healthy tissues.
Despite these innovative biotechnologies with novel mechanisms of tumor killing, chemotherapy remains a pillar of medical oncology. However, the precise elimination of cancer cells by modern targeted and immune therapies begs the question, is it possible to retain the efficacy of chemotherapy while reducing its toxic effects on healthy tissues? In other words, can the therapeutic index of chemotherapy be increased? Multiple approaches have been evaluated to this end. A promising method has been the use of nanoparticle formulations for chemo drugs such as liposomal daunorubicin or nab-paclitaxel [48-50]. With nanoparticles, compounds are encapsulated by “bubbles” of protein or fatty molecules known as lipids. These formulations improve the solubility of these drugs and help to provide a steady release of drug into the bloodstream. Although often superior to traditional chemo, these formulations rely on passive accretion into tumors driven by factors such as nanoparticle size, charge, and tumor permeability [51, 52]. As such, the efficacy of this approach varies substantially depending on the tumor type and its unique vascular architecture. In contrast to passive drug accumulation, other delivery techniques have been engineered to actively target chemotherapy to cancer cells. The most successful has been the invention of antibody-drug conjugates (ADCs).
Antibody-Drug Conjugates
Utilizing an antibody for delivering chemotherapeutic drugs to cancer cells was described as early as 1958 but remained a niche area of research for several decades [53-56]. These immunoglobulin proteins are particularly well-suited as targeting vectors due to their exquisite binding selectivity and long half-life in the bloodstream [57]. As a drug class, antibodies can be developed to specifically recognize almost any protein of interest while circumventing interactions with other nearby biomolecules [58]. With this engineering capability in hand, researchers first work to identify proteins that are expressed on the surface of cancer cells exclusively but are not present on healthy tissues. The cancer-specific proteins behave as a molecular “handle” on the cell surface that can be specifically latched onto by antibodies as they pass by in circulation, and therefore result in an accumulation of therapeutic immunoglobulins inside the tumor over time. An ADC can be broken down into three basic components [59]. 1) the antibody, specially designed for binding a cancer protein of interest, 2) the drug, a molecular “payload” that will be delivered into the cell. Typically, this is a highly toxic chemotherapy or derivative thereof, and 3) the linker, a short chemical tether that physically connects the antibody to the drug. Linkers dictate when and where the payload is released and are highly important for the overall therapeutic performance of an ADC [60]. The enrichment of ADCs within a tumor creates an environment where concentrated doses of toxin are localized to malignant tissues. Consequently, the therapeutic index of these drugs is often higher than systemic chemotherapy [61]. A higher therapeutic index allows for lower levels of toxicity while maintaining the same efficacy or, alternatively, higher efficacy while maintaining equivalent toxicity.
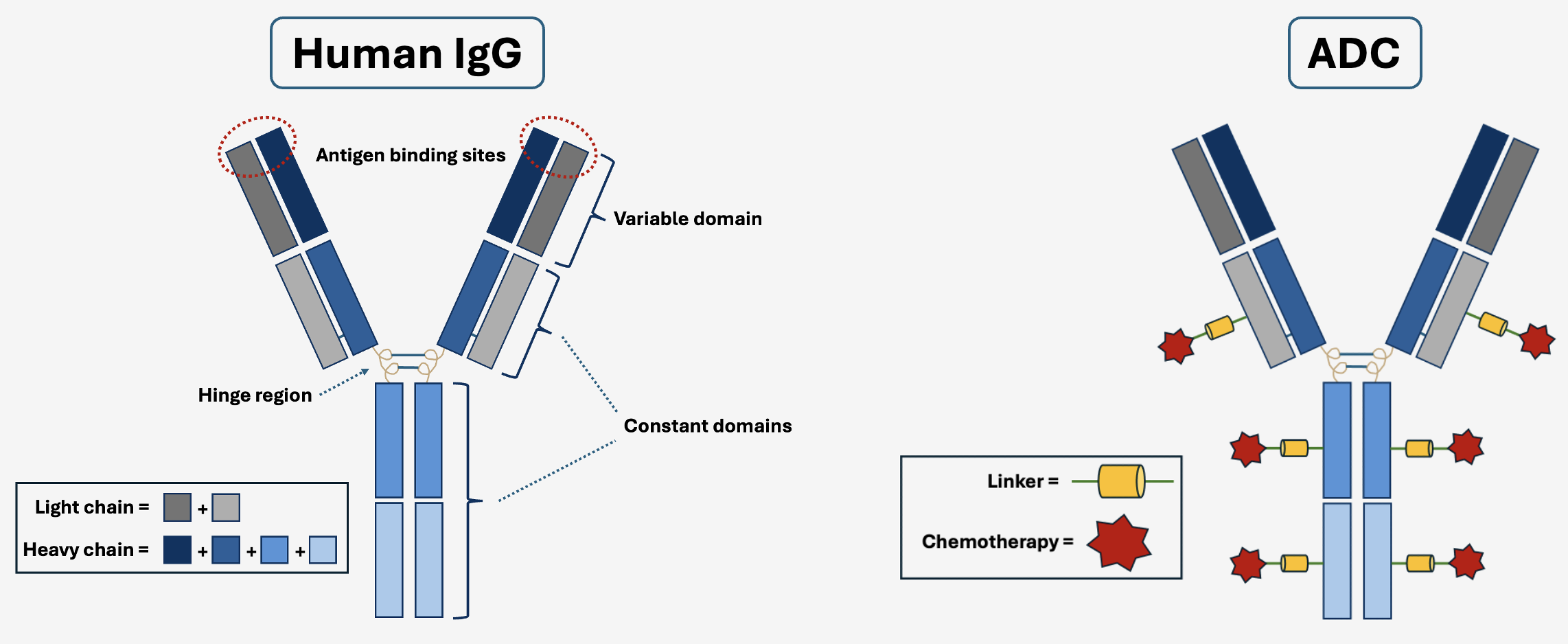
Figure 3. Structure of Human IgG Compared to an ADC. Fusing a chemotherapy to a human antibody (IgG) via a selectively cleavable linker is the most common form of ADC. There are multiple copies of drug molecules on a single antibody, also known as the drug-antibody ratio (DAR).
Once the antibody portion of an ADC is bound to its protein receptor on a cancer cell, it is internalized through a process known as receptor-mediated endocytosis, or just endocytosis for short [62]. During endocytosis, a localized region of the cellular membrane cavitates to form an intracellular vesicle in which receptors that were once located on the cell surface now line the inside of a membranous pouch known as an endosome [63, 64]. Multiple biochemical changes occur within the localized environment of an endosome as it matures, such as a decrease in pH and an increased concentration of enzymes known as proteases that break down endosomal proteins. In the majority of FDA approved ADCs on the market today, the linker moiety connecting the antibody to its toxic chemotherapy payload is specifically engineered to degrade after contacting the unique endosomal milieu [65]. Some linkers are sensitive to the low pH environment, known as “acid labile,” while others are cleaved by endosomal proteases known as cathepsins. Once cleaved, the payload is free to diffuse into the cellular cytoplasm or nucleus to exert its toxic effects. There are a number of factors that influence how much toxin actually reaches its final cellular destination, such as the number of receptor molecules on the cell surface available for antibody binding, how frequently these receptors internalize into endosomes, and the efficiency of payload cleavage [66]. Moreover, ADCs can have varying amounts of drug molecules attached to the antibody, known as the drug-antibody ratio (DAR). The typical DAR ranges from two to eight drug molecules per antibody and strongly influences properties such as efficacy and antibody stability [66-68].

Figure 4. ADCs Specifically Target Cancer Cells. The antibody portion of the ADC binds to cell-surface proteins uniquely expressed on cancerous tissues, enabling precise delivery to malignant cells while sparing healthy counterparts.
The selective release of drug upon entering the endosome is vital for maximizing safety and efficacy of an ADC. Toxic payloads are not able to penetrate healthy cells while they are still attached to an ADC, as antibodies cannot passively diffuse across a cell membrane and hence require internalization by receptor-mediated endocytosis. Once cleaved from the ADC, many payloads are able to traverse membranes due to their small and hydrophobic nature. If a linker is not sufficiently stable in the bloodstream, then cleavage may happen prematurely before reaching the endosome within a cancer cell. When this occurs, drug is released from the ADC and will diffuse into any cell in close proximity to induce apoptosis, regardless of whether it is healthy or cancerous. This is obviously an undesirable outcome and will result in adverse side effects for patients. Linker instability was a major problem for early versions of ADCs and took years of research and development to determine the optimal chemical architectures for specific endosomal release [69, 70]. Innovation in linker technology continues to evolve towards greater stability in the blood and specific release only in the cell type of interest.
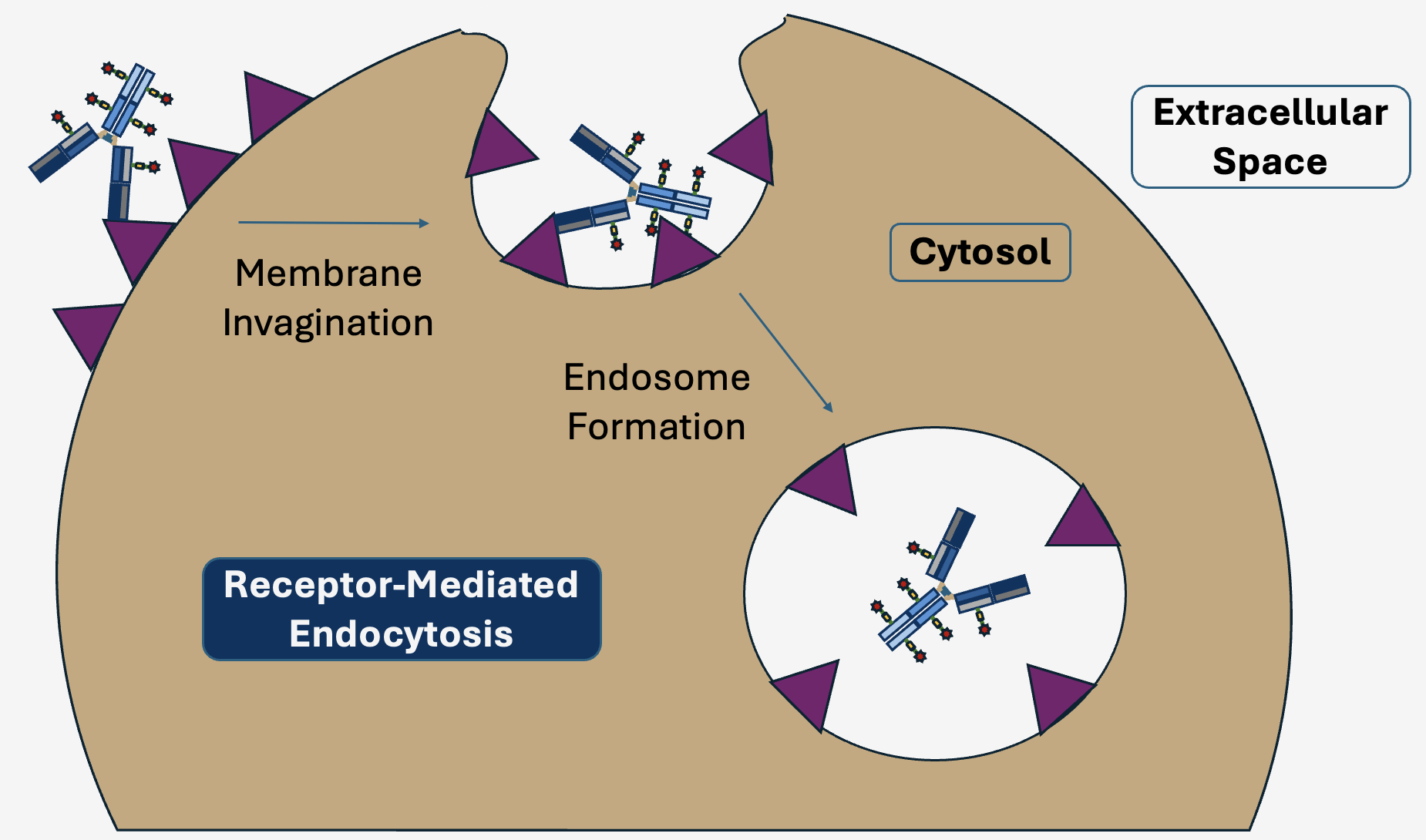
Figure 5. ADC Uptake by Endocytosis. Upon binding its specific cell-surface receptor, an ADC is internalized via receptor mediated-endocytosis into an endosome.
Choosing the optimal cytotoxic payload is also a key aspect of ADC design. Initial studies revealed that the ideal toxin needs to be extremely potent, as ADCs crafted using traditional chemotherapeutic drugs were not powerful enough to sufficiently eliminate tumors [66]. In fact, payloads being used today are 10-100x more lethal to cells than typical systemic chemotherapies [66]. Although more potent, modern payloads are similar to traditional chemo in their mechanism of cell killing. Some popular choices have been anti-mitotic drugs such as MMAE, DM1, and DM4 or topoisomerase inhibitors such as DXd and SN-38 [70]. In addition to their toxicity, physiochemical attributes of the payload are also important characteristics to consider. As mentioned above, once a drug has been released from the ADC it is able to freely diffuse around its local environment. If the payload is sufficiently hydrophobic, it may pass through the cellular membrane and penetrate neighboring cells. This characteristic is a double-edged sword. If an ADC is floating through bloodstream, prematurely discharging toxin into healthy tissue is clearly a bad thing. In this case, using a hydrophilic toxin that cannot permeate the membranes of healthy cells would be beneficial. However, once inside of a tumor the ability of a payload to cross membranes can be advantageous, as neighboring cells are likely also cancerous. This is known as the “bystander effect” and is increasingly recognized as an important property for effective tumor destruction [71, 72]. Tumors are notoriously heterogeneous, containing various sub-populations of cancer cells within them that each have their own unique molecular fingerprints. As a result, ADCs may successfully bind populations of cancer cells within a tumor that express the target protein on its surface, but pass by others that do not. The bystander effect helps address this issue and ensures a more complete elimination of all surrounding cancer cells.
The Promise of Modern ADCs
As a biotechnology, it took decades of focused research by scientists and physicians before ADCs reached the necessary maturity to be clinically viable. Fortunately, these efforts have paid off handsomely – the efficacy and safety of contemporary ADCs has been met with global renown throughout the medical community. Headlines expound standing ovations from crowds of physician scientists at medical conferences where exceptional new data is first revealed to the public [73, 74]. There are currently 11 ADCs approved by the FDA for use across many types of cancer [75-85]. Padcev and Enhertu, two of the fastest growing ADCs used in clinical practice today [86], are great examples of how these new medicines are changing the lives of patients.

Figure 6. Endosomal Release of Toxin Payload From an ADC. Confined within the endosome, proteases and a low pH environment work in tandem to cleave the ADC linker moiety and degrade the antibody. This frees the chemotherapy to diffuse into the cytosol or nucleus and exert its lethal effects.
Padcev is an ADC co-marketed by Pfizer and Astellas composed of a fully human antibody specifically targeting the cancer-associated protein Nectin-4, fused to the microtubule inhibitor MMAE by a selectively cleavable di-peptide linkage [87]. Nectin-4 was shown to be highly expressed in bladder cancer, and hence is an ideal protein target for this specific indication [87]. The drug is approved for use in adults with locally advanced or metastatic urothelial cancer (advanced bladder cancer) in combination with the PD-1 immune checkpoint inhibitor pembrolizumab (Keytruda, Merck) [81]. Padcev made waves among physicians after data from the clinical trial EV-302 was presented in 2023 at the prominent medical conference ESMO. When interpreting clinical data from novel cancer therapeutics, arguably the most important factor to analyze is overall survival (OS). This metric demonstrates the likelihood of a patient remaining alive after a certain period of time, comparing those who receive the experimental drug versus those treated with the current standard of care. Increasing OS is extremely important clinically as well as from an insurance coverage and reimbursement perspective. The EV-302 trial showed that in previously untreated patients receiving a combination of Padcev and Keytruda, the risk of death was cut in half compared to those receiving traditional platinum-based chemotherapy (median OS of 31.5 months vs 16.1 months) [88]. In other words, an average patient on the Padcev plus Keytruda combo was expected to live twice as long relative to those on standard chemo. An efficacy improvement of this magnitude is quite extraordinary.
Let’s imagine from the patient perspective how this impacts their journey of care. A person visits their doctor and learns they have metastatic bladder cancer. As a first line of defense, the oncologist decides to initiate treatment with Padcev and Keytruda. Based on the EV-302 data, there is a ~75% chance the patient will still be alive one year later, a ~60% chance two years later, and a ~35% chance three years later. Now assume an alternative scenario where that same patient received traditional platinum-based chemo instead of Padcev and Keytruda. Their probability of survival is ~60% after one year, ~35% after two years, and ~25% after three years. Clearly, increasing the odds of living for two years from ~35% to ~60% is a tremendous benefit. Not only are ADCs replacing chemotherapy as the first line of treatment for some cancer types, they are also offering hope for patients with relapsing disease after receiving other medicines. In fact, newer generations of ADCs are already beginning to outperform older ADC drugs in clinical trials. Enhertu is one of these with an outstanding clinical profile.

Figure 7. List of Currently FDA Approved ADCs. The medicines treat a wide range of liquid and solid tumors. * = following market withdrawal in 2010, Mylotarg was FDA approved in 2017 using a modified dose.
Enhertu is an ADC co-marketed by Daiichi-Sankyo and AstraZeneca for the treatment of HER2-positive metastatic breast cancer [80]. Its constituent pieces are a humanized antibody directed towards the protein HER2 and the topoisomerase inhibitor DXd, connected together by a protease-cleavable tetra-peptide linker [80]. In the clinical trial DESTINY-Breast03 [89], patients were enrolled who had metastatic breast cancer and previously failed treatment with chemotherapy (taxanes) plus the anti-HER2 antibody trastuzumab (Herceptin, Roche). Patients were randomly assigned to receive either Enhertu or an earlier HER2-ADC known as Kadcyla (Roche). An important detail is that both Enhertu and Kadcyla utilize the same antibody as Herceptin (trastuzumab), but are differentiated by their linker and toxic payload. Patients who received Enhertu had a greater OS compared to those on Kadcyla (mOS of 40.5 months vs. 34.0 months) [89], demonstrating the superiority of this medicine to its older generation counterpart. Furthermore, another clinical trial (DESTINY-Breast02) [90] evaluated the performance of Enhertu versus chemotherapy (capecitabine) plus Herceptin or lapatinib (Tykerb, GSK) in patients who were previously treated with Kadcyla and experienced disease progression. Those on Enhertu displayed a mOS of 39.2 months, while the chemotherapy combination groups had a mOS of 26.5 months [90]. Taken together, these data show the tremendous benefits of Enhertu and position this drug as an important treatment option for patients as both a replacement for older ADCs and as a second choice after disease progression on other therapies.
Limitations
While ADCs have delivered outstanding results to patients, they are not infallible. These medicines are more selective than traditional chemotherapy, but each have their own set of drawbacks. Side effects can be broadly grouped into two categories – “on-target” toxicities, and “off-target” toxicities. As the name implies, “off-target” toxicities refer to side effects that are caused by a drug exerting its effect outside of its intended molecular destination. In the case of ADCs, there are a few typical ways in which this can happen. The most common is premature release of toxin payload in the bloodstream before reaching the tumor [91]. Although linkers are engineered to rapidly discharge payload within the endosome of a cancer cell, they also slowly degrade over time when circulating throughout the body and thus toxin is free to damage healthy cells. Researchers are constantly searching for new linker technologies that enhance stability in order to increase safety and efficacy. Furthermore, toxicity profiles have been strongly associated with the identity of payload fused to an ADC [92]. As such, large investments are being made to identify chemotoxins with better tolerability while maintaining efficacy. Another off-target mechanism is the uptake of ADCs into immune cells through a cell surface protein known as the Fc receptor [91]. Fc receptor-mediated internalization of antibodies by white blood cells is part of their normal functioning in the body, but is obviously undesirable for an ADC and can result in side effects such as neutropenia and thrombocytopenia. To prevent this unwanted uptake, companies have developed special classes of antibodies harboring mutations that disrupt their interaction with Fc-receptors and thus reduce internalization [93].
In the category of “on-target” toxicities, these side effects are fundamentally related to the protein in which an ADC is designed to target. An ideal cancer-protein target is one that shows high levels of expression on cancer cells while no expression on healthy tissues. In practice, a protein target with this characteristic is exceedingly difficult to find. Most often there is a strong enrichment of a protein in a cancer cell relative to normal tissue, but there still remains low levels of expression on some healthy cells. As a result, the ADC will predominantly localize to the tumor, yet also bind to normal tissue at a lesser extent. Every ADC will have a different “on-target” toxicity profile depending on which cancer-associated protein the antibody is intended to recognize and its expression levels across different tissues in the body. For example, Padcev is known to cause significant rates of dysgeusia, meaning a persistent fowl or unpleasant taste in the mouth [81]. This side effect is thought to result from appreciable expression of the target protein Nectin-4 in salivary glands [87]. Researchers are perpetually on the hunt for better, cancer-specific targets as the next generation of ADCs are in development.
The Future
Superb clinical data has fueled a resurgence of interest throughout the entire biotechnology industry. Many of the largest pharmaceutical companies have now entered the treatment space and a wide range of clinical trials are ongoing. There are numerous unique approaches being tested clinically, encompassing a tremendous variety of cancers. Exploring new types of cancer protein targets, linker chemistries, and toxic payloads have created the potential for drugs with even greater efficacy and safety, as well as potentially generating new therapies in areas of high unmet need. Additionally, some organizations are investigating the delivery of medicines outside of chemotherapy altogether. One such focus is on creating ADCs with immune-stimulating drugs known as toll-like receptor (TLR) agonists [94, 95]. Similar to PD-1 inhibitors, the hope is that combining an immune-activating TLR-ADC with a traditional, toxin-linked ADC will supercharge the efficacy of these drugs. Groups are also exploring ADCs in the field of genetic medicine to deliver large biomolecule drugs such as antisense oligonucleotides (ASOs) and small interfering RNAs (siRNAs) [96]. A major issue hindering development of these nucleic acid therapeutics has been the targeted delivery to cell types of interest. The exquisite binding specificity of antibodies has inspired development of ADCs as a molecular shuttle for ASOs and siRNAs. ADCs may appear to have burst on the medical scene only recently, but this technology has been many decades in the making. The future is particularly exciting, as more novel candidates have entered the clinic than ever before. A tremendous collective investment among biotechnology and pharmaceutical companies has fueled an explosion of innovation that will ultimately benefit patients around the globe. The next 10 years and beyond are looking very bright for this rising star modality.
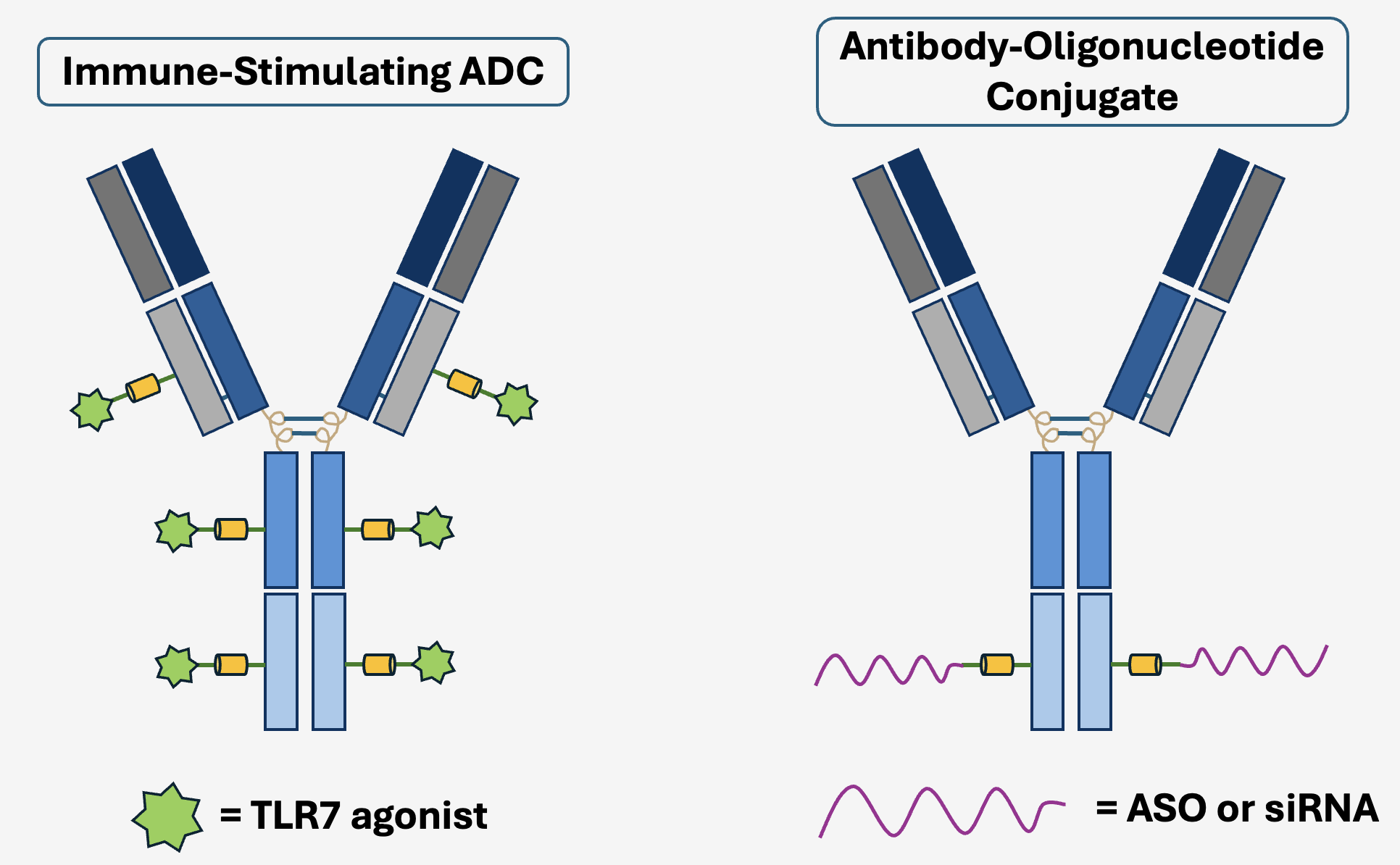
Figure 8. Novel ADC Formats. Exploratory payloads include immune stimulating compounds such as TLR agonists or nucleic acid therapeutics such as ASOs.
Supplemental Figures
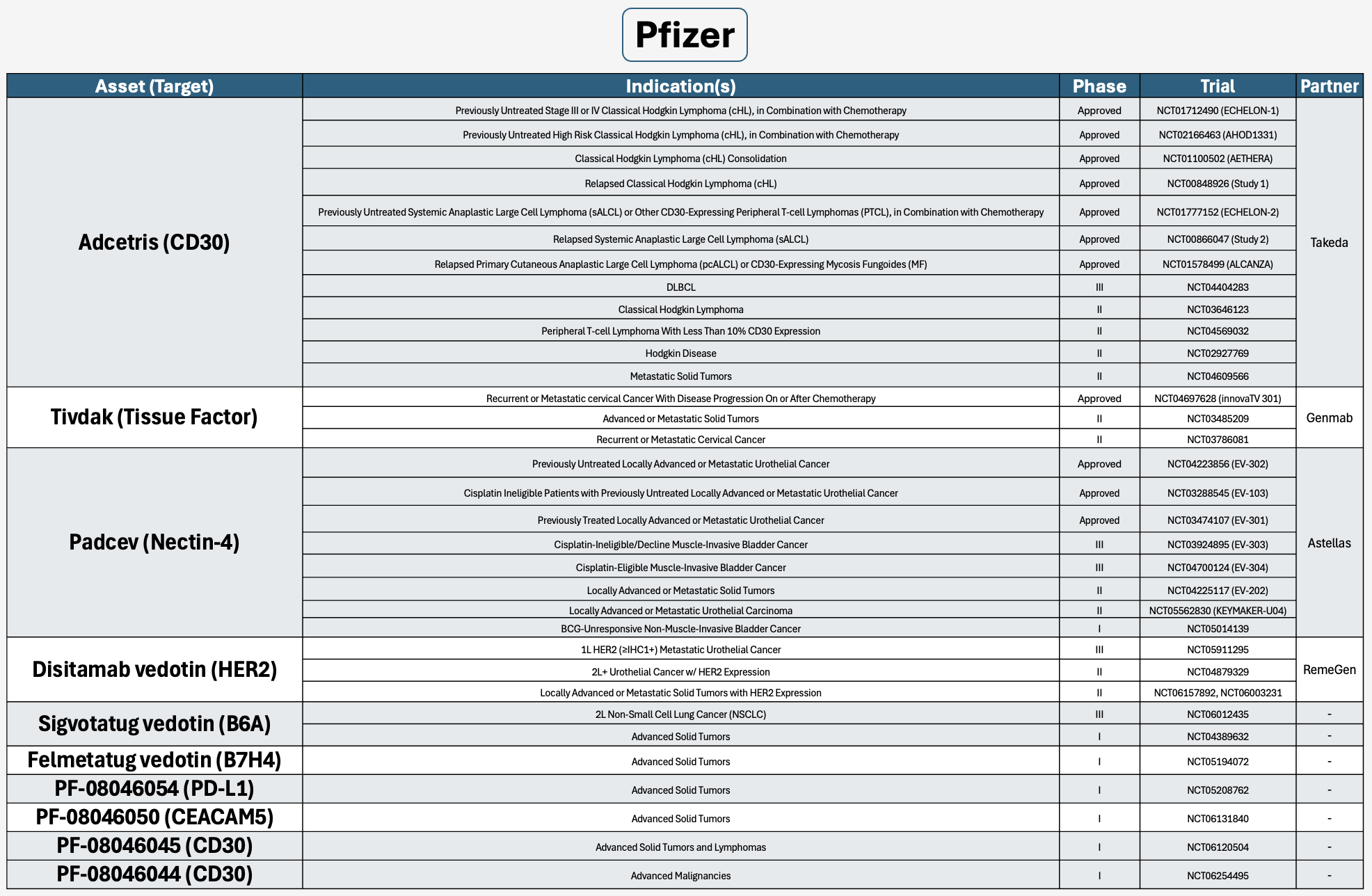
Major companies with at least 5 ADCs in development.




Conflicts of Interest
At the time of writing, JMH has beneficial equity positions in the following companies mentioned in this article: Pfizer.
References
1. Wyld, L., R.A. Audisio, and G.J. Poston, The evolution of cancer surgery and future perspectives.Nat Rev Clin Oncol, 2015. 12(2): p. 115-24.
2. Fidler, I.J., The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer, 2003. 3(6): p. 453-8.
3. UK, C.R. What is cancer surgery? 2022; Available from: https://www.cancerresearchuk.org/about-cancer/treatment/surgery/about.
4. Beddok, A., et al., A Comprehensive Primer on Radiation Oncology for Non-Radiation Oncologists. Cancers (Basel), 2023. 15(20).
5. Early Breast Cancer Trialists' Collaborative, G., et al., Effect of radiotherapy after breast-conserving surgery on 10-year recurrence and 15-year breast cancer death: meta-analysis of individual patient data for 10,801 women in 17 randomised trials. Lancet, 2011. 378(9804): p. 1707-16.
6. Amjad, M.T., A. Chidharla, and A. Kasi, Cancer Chemotherapy, in StatPearls. 2024: Treasure Island (FL).
7. Cooper, G., The Cell: A Molecular Approach. 2nd ed. Cell Proliferation in Development and Differentiation. 2000, Sunderland (MA): Sinauer Associates.
8. Mitchison, T.J., The proliferation rate paradox in antimitotic chemotherapy. Mol Biol Cell, 2012. 23(1): p. 1-6.
9. McIntosh, J.R., Mitosis. Cold Spring Harb Perspect Biol, 2016. 8(9).
10. Song, H.Y., et al., DNA replication: Mechanisms and therapeutic interventions for diseases. MedComm (2020), 2023. 4(1): p. e210.
11. Chatterjee, N. and G.C. Walker, Mechanisms of DNA damage, repair, and mutagenesis. Environ Mol Mutagen, 2017. 58(5): p. 235-263.
12. Berti, M., D. Cortez, and M. Lopes, The plasticity of DNA replication forks in response to clinically relevant genotoxic stress. Nat Rev Mol Cell Biol, 2020. 21(10): p. 633-651.
13. Roos, W.P. and B. Kaina, DNA damage-induced cell death by apoptosis. Trends Mol Med, 2006. 12(9): p. 440-50.
14. DeVita, V.T., Jr. and E. Chu, A history of cancer chemotherapy. Cancer Res, 2008. 68(21): p. 8643-53.
15. National Cancer Institute. Milestones in Cancer Research and Discovery. August 31st, 2020; Available from: https://www.cancer.gov/research/progress/250-years-milestones.
16. Krumbhaar, E.B. and H.D. Krumbhaar, The Blood and Bone Marrow in Yelloe Cross Gas (Mustard Gas) Poisoning: Changes produced in the Bone Marrow of Fatal Cases. J Med Res, 1919. 40(3): p. 497-508 3.
17. Goodman, L.S., et al., Landmark article Sept. 21, 1946: Nitrogen mustard therapy. Use of methyl-bis(beta-chloroethyl)amine hydrochloride and tris(beta-chloroethyl)amine hydrochloride for Hodgkin's disease, lymphosarcoma, leukemia and certain allied and miscellaneous disorders. By Louis S. Goodman, Maxwell M. Wintrobe, William Dameshek, Morton J. Goodman, Alfred Gilman and Margaret T. McLennan. JAMA, 1984. 251(17): p. 2255-61.
18. Ogino, M.H. and P. Tadi, Cyclophosphamide, in StatPearls. 2024: Treasure Island (FL).
19. Gold, J.M. and A. Raja, Cisplatin, in StatPearls. 2024: Treasure Island (FL).
20. Devanabanda, B. and A. Kasi, Oxaliplatin, in StatPearls. 2024: Treasure Island (FL).
21. Casale, J. and P. Patel, Fluorouracil, in StatPearls. 2024: Treasure Island (FL).
22. Sharma, H. and R. Wadhwa, Mercaptopurine, in StatPearls. 2024: Treasure Island (FL).
23. National Cancer Institute. Gemcitabine Hydrochloride. September 14th, 2023; Available from: https://www.cancer.gov/about-cancer/treatment/drugs/gemcitabinehydrochloride.
24. Johnson-Arbor, K. and R. Dubey, Doxorubicin, in StatPearls. 2024: Treasure Island (FL).
25. Reyhanoglu, G. and P. Tadi, Etoposide, in StatPearls. 2024: Treasure Island (FL).
26. Reyhanoglu, G. and T. Smith, Irinotecan, in StatPearls. 2024: Treasure Island (FL).
27. Awosika, A.O., M.C. Farrar, and T.F. Jacobs, Paclitaxel, in StatPearls. 2024: Treasure Island (FL).
28. Farha, N.G. and A. Kasi, Docetaxel, in StatPearls. 2024: Treasure Island (FL).
29. National Cancer Institute. Vinblastine Sulfate. October 4th, 2023; Available from: https://www.cancer.gov/about-cancer/treatment/drugs/vinblastinesulfate.
30. Scharer, O.D., Chemistry and biology of DNA repair. Angew Chem Int Ed Engl, 2003. 42(26): p. 2946-74.
31. Li, L.Y., et al., DNA Repair Pathways in Cancer Therapy and Resistance. Front Pharmacol, 2020. 11: p. 629266.
32. Parker, W.B., Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer. Chem Rev, 2009. 109(7): p. 2880-93.
33. Pommier, Y., et al., Human topoisomerases and their roles in genome stability and organization. Nat Rev Mol Cell Biol, 2022. 23(6): p. 407-427.
34. Cheng, Z., X. Lu, and B. Feng, A review of research progress of antitumor drugs based on tubulin targets. Transl Cancer Res, 2020. 9(6): p. 4020-4027.
35. Florian, S. and T.J. Mitchison, Anti-Microtubule Drugs. Methods Mol Biol, 2016. 1413: p. 403-21.
36. Bowater, R.J., S.M. Abdelmalik, and R.J. Lilford, Efficacy of adjuvant chemotherapy after surgery when considered over all cancer types: a synthesis of meta-analyses. Ann Surg Oncol, 2012. 19(11): p. 3343-50.
37. Anampa, J., D. Makower, and J.A. Sparano, Progress in adjuvant chemotherapy for breast cancer: an overview.BMC Med, 2015. 13: p. 195.
38. Chan, G.H.J. and C.E. Chee, Making sense of adjuvant chemotherapy in colorectal cancer. J Gastrointest Oncol, 2019. 10(6): p. 1183-1192.
39. Alam, N., et al., Adjuvant chemotherapy for completely resected non-small cell lung cancer: a systematic review.Crit Rev Oncol Hematol, 2006. 58(2): p. 146-55.
40. Tilsed, C.M., et al., Cancer chemotherapy: insights into cellular and tumor microenvironmental mechanisms of action. Front Oncol, 2022. 12: p. 960317.
41. Muller, P.Y. and M.N. Milton, The determination and interpretation of the therapeutic index in drug development. Nat Rev Drug Discov, 2012. 11(10): p. 751-61.
42. Gerber, D.E., Targeted therapies: a new generation of cancer treatments. Am Fam Physician, 2008. 77(3): p. 311-9.
43. Bedard, P.L., et al., Small molecules, big impact: 20 years of targeted therapy in oncology. Lancet, 2020. 395(10229): p. 1078-1088.
44. Lackner, M.R., T.R. Wilson, and J. Settleman, Mechanisms of acquired resistance to targeted cancer therapies.Future Oncol, 2012. 8(8): p. 999-1014.
45. Waldman, A.D., J.M. Fritz, and M.J. Lenardo, A guide to cancer immunotherapy: from T cell basic science to clinical practice. Nat Rev Immunol, 2020. 20(11): p. 651-668.
46. Balar, A.V. and J.S. Weber, PD-1 and PD-L1 antibodies in cancer: current status and future directions. Cancer Immunol Immunother, 2017. 66(5): p. 551-564.
47. Bonaventura, P., et al., Cold Tumors: A Therapeutic Challenge for Immunotherapy. Front Immunol, 2019. 10: p. 168.
48. Wicki, A., et al., Nanomedicine in cancer therapy: challenges, opportunities, and clinical applications. J Control Release, 2015. 200: p. 138-57.
49. Stinchcombe, T.E., Nanoparticle albumin-bound paclitaxel: a novel Cremphor-EL-free formulation of paclitaxel. Nanomedicine (Lond), 2007. 2(4): p. 415-23.
50. Dawidczyk, C.M., et al., Tumor accumulation of liposomal doxorubicin in three murine models: Optimizing delivery efficiency. Nanomedicine, 2017. 13(5): p. 1637-1644.
51. Giri, P.M., A. Banerjee, and B. Layek, A Recent Review on Cancer Nanomedicine. Cancers (Basel), 2023. 15(8).
52. Wong, A.D., et al., Quantitative Analysis of the Enhanced Permeation and Retention (EPR) Effect. PLoS One, 2015. 10(5): p. e0123461.
53. Mathe, G., L.O. Tran Ba, and J. Bernard, [Effect on mouse leukemia 1210 of a combination by diazo-reaction of amethopterin and gamma-globulins from hamsters inoculated with such leukemia by heterografts]. C R Hebd Seances Acad Sci, 1958. 246(10): p. 1626-8.
54. Ghose, T., et al., Immunochemotherapy of cancer with chlorambucil-carrying antibody. Br Med J, 1972. 3(5825): p. 495-9.
55. Ghose, T. and A.H. Blair, Antibody-linked cytotoxic agents in the treatment of cancer: current status and future prospects. J Natl Cancer Inst, 1978. 61(3): p. 657-76.
56. Ford, C.H., et al., Localisation and toxicity study of a vindesine-anti-CEA conjugate in patients with advanced cancer. Br J Cancer, 1983. 47(1): p. 35-42.
57. Ovacik, M. and K. Lin, Tutorial on Monoclonal Antibody Pharmacokinetics and Its Considerations in Early Development. Clin Transl Sci, 2018. 11(6): p. 540-552.
58. Sharma, P., et al., Therapeutic Antibodies in Medicine. Molecules, 2023. 28(18).
59. Fu, Z., et al., Antibody drug conjugate: the "biological missile" for targeted cancer therapy. Signal Transduct Target Ther, 2022. 7(1): p. 93.
60. Nolting, B., Linker technologies for antibody-drug conjugates. Methods Mol Biol, 2013. 1045: p. 71-100.
61. Gerber, H.P., S. Gangwar, and A. Betts, Therapeutic index improvement of antibody-drug conjugates. MAbs, 2023. 15(1): p. 2230618.
62. Chalouni, C. and S. Doll, Fate of Antibody-Drug Conjugates in Cancer Cells. J Exp Clin Cancer Res, 2018. 37(1): p. 20.
63. Wileman, T., C. Harding, and P. Stahl, Receptor-mediated endocytosis. Biochem J, 1985. 232(1): p. 1-14.
64. Doherty, G.J. and H.T. McMahon, Mechanisms of endocytosis. Annu Rev Biochem, 2009. 78: p. 857-902.
65. Gogia, P., et al., Antibody-Drug Conjugates: A Review of Approved Drugs and Their Clinical Level of Evidence.Cancers (Basel), 2023. 15(15).
66. Nakada, T., et al., The Latest Research and Development into the Antibody-Drug Conjugate, [fam-] Trastuzumab Deruxtecan (DS-8201a), for HER2 Cancer Therapy. Chem Pharm Bull (Tokyo), 2019. 67(3): p. 173-185.
67. Sun, X., et al., Effects of Drug-Antibody Ratio on Pharmacokinetics, Biodistribution, Efficacy, and Tolerability of Antibody-Maytansinoid Conjugates. Bioconjug Chem, 2017. 28(5): p. 1371-1381.
68. Lyon, R.P., et al., Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat Biotechnol, 2015. 33(7): p. 733-5.
69. Joubert, N., et al., Antibody-Drug Conjugates: The Last Decade. Pharmaceuticals (Basel), 2020. 13(9).
70. Baah, S., M. Laws, and K.M. Rahman, Antibody-Drug Conjugates-A Tutorial Review. Molecules, 2021. 26(10).
71. Giugliano, F., et al., Bystander effect of antibody-drug conjugates: fact or fiction? Curr Oncol Rep, 2022. 24(7): p. 809-817.
72. Li, F., et al., Intracellular Released Payload Influences Potency and Bystander-Killing Effects of Antibody-Drug Conjugates in Preclinical Models. Cancer Res, 2016. 76(9): p. 2710-9.
73. Ledford, H., Cancer trial results show power of weaponized antibodies. Nature, 2023. 623(7986): p. 231-232.
74. Ben-Ari, E. Enhertu Improves Survival for Metastatic “HER2-Low” Breast Cancer. 2022.
75. Pfizer. Mylotarg Prescribing Information. 2021; Available from: https://labeling.pfizer.com/ShowLabeling.aspx?id=9548.
76. Pfizer. Adcetris Prescribing Information. 2023; Available from: https://docs.seagen.com/Adcetris_Full_Ltr_Master.pdf.
77. Roche. Kadcyla Prescribing Information. 2022; Available from: https://www.gene.com/download/pdf/kadcyla_prescribing.pdf.
78. Pfizer. Besponsa Prescribing Information. 2024; Available from: https://labeling.pfizer.com/ShowLabeling.aspx?id=9503.
79. Roche. Polivy Prescribing Information. 2023; Available from: https://www.gene.com/download/pdf/polivy_prescribing.pdf.
80. Daiichi-Sankyo. Enhertu Prescribing Information. 2024; Available from: https://daiichisankyo.us/prescribing-information-portlet/getPIContent?productName=Enhertu&inline=true.
81. Astellas. Padcev Prescribing Information. 2023; Available from: https://astellas.us/docs/PADCEV_label.pdf.
82. Gilead. Trodelvy Prescribing Information. 2024; Available from: https://www.gilead.com/-/media/files/pdfs/medicines/oncology/trodelvy/trodelvy_pi.pdf.
83. Therapeutics, A. Zynlonta Prescribing Information. 2022; Available from: https://www.adctherapeutics.com/wp-content/uploads/2023/10/ZYNLONTA-PI_October-2022_LOCKED.pdf.
84. Pfizer. Tivdak Prescribing Information. 2024; Available from: https://docs.seagen.com/Tivdak_Full_Ltr_Master.pdf.
85. AbbVie. Elahere Prescribing Information. 2024; Available from: https://elahere.com/pdf/prescribing-information.pdf.
86. do Pazo, C., K. Nawaz, and R.M. Webster, The oncology market for antibody-drug conjugates. Nat Rev Drug Discov, 2021. 20(8): p. 583-584.
87. Challita-Eid, P.M., et al., Enfortumab Vedotin Antibody-Drug Conjugate Targeting Nectin-4 Is a Highly Potent Therapeutic Agent in Multiple Preclinical Cancer Models. Cancer Res, 2016. 76(10): p. 3003-13.
88. Powles, T., et al., Enfortumab Vedotin and Pembrolizumab in Untreated Advanced Urothelial Cancer. N Engl J Med, 2024. 390(10): p. 875-888.
89. Cortes, J., et al., Trastuzumab Deruxtecan versus Trastuzumab Emtansine for Breast Cancer. N Engl J Med, 2022. 386(12): p. 1143-1154.
90. Andre, F., et al., Trastuzumab deruxtecan versus treatment of physician's choice in patients with HER2-positive metastatic breast cancer (DESTINY-Breast02): a randomised, open-label, multicentre, phase 3 trial. Lancet, 2023. 401(10390): p. 1773-1785.
91. Nguyen, T.D., B.M. Bordeau, and J.P. Balthasar, Mechanisms of ADC Toxicity and Strategies to Increase ADC Tolerability. Cancers (Basel), 2023. 15(3).
92. Colombo, R. and J.R. Rich, The therapeutic window of antibody drug conjugates: A dogma in need of revision.Cancer Cell, 2022. 40(11): p. 1255-1263.
93. Wilkinson, I., et al., Fc-engineered antibodies with immune effector functions completely abolished. PLoS One, 2021. 16(12): p. e0260954.
94. Pfizer, Oncology Innovation Day. 2024.
95. Mills, D. et al. Abstract 6352: Preclinical discovery of ARX622: A site-specific, TLR7 agonist, HER2-targeted immune-stimulatory antibody drug conjugate for treatment of multiple solid tumor types. 2024; Available from: https://aacrjournals.org/cancerres/article/84/6_Supplement/6352/740488/Abstract-6352-Preclinical-discovery-of-ARX622-A.
96. Mullard, A., Antibody-oligonucleotide conjugates enter the clinic. Nat Rev Drug Discov, 2022. 21(1): p. 6-8.